




La Sindrome di Alström
di Pietro MaffeiDefinizione
Quadro clinico
Apparato visivo. Apparato uditivo. Sistema endocrino-metabolico. Apparato cardiocircolatorio. Apparato urinario. Sistema gastrointestinale. Sistema nervoso centrale.Genetica
Patologia
Diagnosi
Terapia
Bibliografia essenziale
Definizione
La sindrome di Alström (ALMS) è una rara malattia autosomica recessiva. Si caratterizza per la comparsa progressiva di obesità e diabete mellito di tipo II, degenerazione retinica ed ipoacusia. Nel 1997 è stato individuato il gene responsabile della sindrome (ALMS1, cr. 2p13) e da allora è possibile una diagnosi genetica a conferma della diagnosi clinica. Finora sono stati descritti circa 300 pazienti con ALMS nel mondo.

Quadro clinico
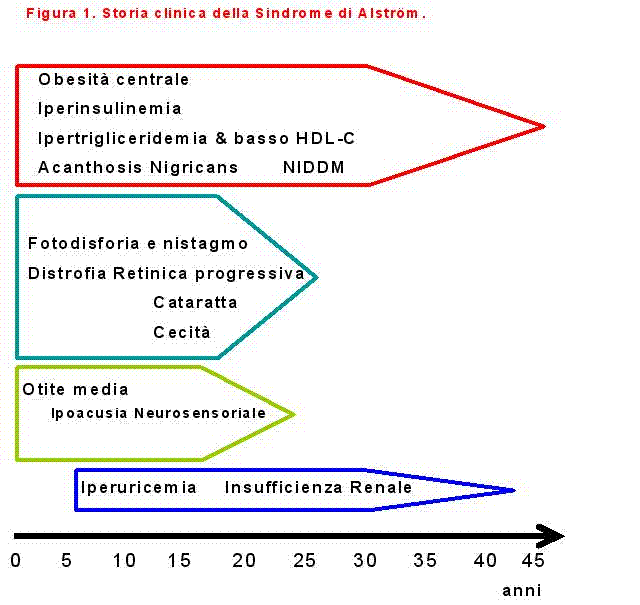
La distrofia retinica compare molto precocemente, solitamente già nel primo anno di vita, e la retinopatia pigmentosa è un aspetto distintivo dell’ALMS. Fra i disturbi visivi iniziali ricordiamo il nistagmo che compare precocemente già nei primi mesi di vita, il blefarospasmo secondario a fotofobia, e una riduzione dell’acuità visiva. Nella prima decade di vita si osserva una progressiva perdita della vista che conduce solitamente a cecità, sino alla perdita della percezione della luce, nella seconda decade. Un altro aspetto tipico del coinvolgimento oculare nell’ALMS è dato dalla comparsa di cataratta sottocapsulare posteriore. La fluorangiografia mostra un’atrofia epiteliale pigmentosa della retina confluente, severa e diffusa. Il nistagmo tende generalmente a diminuire con il passare degli anni e talvolta scompare completamente. La degenerazione retinica si caratterizza inoltre per la precoce perdita della visione centrale.
E’ presente ipoacusia di tipo neurosensoriale, progressiva e bilaterale. I primi sintomi di ipoacusia sono caratterizzati dalla comparsa di tinniti, acufeni e il paziente perde progressivamente la capacità di comprendere le parole. L’audiogramma mostra generalmente un peggioramento progressivo e simmetrico della percezione dei suoni sia per le alte che per le basse frequenze. L’impiego di protesi acustiche permette tuttavia di correggere il difetto dell’udito nella maggior parte dei casi.
Una caratteristica comune a molti pazienti con ALMS è data dal riscontro precoce di iperinsulinemia, secondaria a resistenza periferica, che nel corso degli anni porta al diabete mellito di tipo 2. Le basi molecolari dell’insulino-resistenza nell’ALMS non sono state ancora chiarite. I pazienti ALMS diabetici tendono ad essere maggiormente responsivi alla terapia con ipoglicemizzanti orali piuttosto che all’insulina e quando questa è necessaria si rendono necessari dosaggi molto elevati per ottenere un buon controllo metabolico.
Nell’ALMS sono state segnalate alterazioni del metabolismo lipidico e in particolare elevati livelli di trigliceridi non sempre correlabili al grado di insulino-resistenza o diabete.
La bassa statura è una caratteristica tipica dei pazienti adulti mentre nei bambini si osserva frequentemente un’altezza normale o superiore alla media. Sempre nei bambini è riscontrabile un’età ossea avanzata. Esistono inoltre sporadiche segnalazioni che evidenziano un deficit di ormone della crescita nell’ALMS.
Nei maschi è presente un ipogonadismo ipergonadotropo, con elevati valori basali di LH-FSH e riduzione dei livelli di testosterone. Talvolta le donne presentano irregolarità mestruali e iperandrogenismo, ma non sono stati riportati casi di ipogonadismo. Fino ad oggi nessun paziente con ALMS ha avuto figli. Una certa percentuale di pazienti sembra essere inoltre affetta da ipotiroidismo non autoimmune.
L’obesità è una caratteristica peculiare e costante dell’età pediatrica, ma la sua eziologia è del tutto sconosciuta. Il peso alla nascita nei pazienti con ALMS è tuttavia normale, per aumentare progressivamente nella prima-seconda decade di vita. A partire dalla seconda-terza decade si assiste generalmente alla normalizzazione del peso.
La cardiomiopatia dilatativa è frequentemente osservabile nei pazienti con ALMS ed è spesso causa di decesso. Il rilievo istopatologico più importante è la marcata fibrosi del tessuto cardiaco. Malgrado la fibrosi sia una caratteristica peculiare della malattia non è stata tuttavia mai documentata un’aumentata prevalenza di aritmie.
I pazienti con ALMS presentano spesso, soprattutto dopo la seconda-terza decade, un progressivo peggioramento della funzione renale caratterizzata da una grave nefroangiosclerosi. L’insufficienza renale può condizionare fortemente la prognosi a lungo termine dei pazienti.
I pazienti affetti da ALMS presentano spesso segni clinici e laboratoristici di coinvolgimento epatico con aumento delle transaminasi. I meccanismi patogenetici del danno epatico sono sconosciuti e determinano uno spettro assai ampio di lesioni che vanno dalla semplice steatosi fino a quadri di cirrosi conclamata con ipertensione portale. L’insufficienza epatica costituisce una causa di decesso relativamente frequente. In alcuni pazienti è riscontrabile reflusso gastroesofageo. Raramente si osservano pancreatici acute, la cui causa sembra da ascrivere all’ipertrigliceridemia.
La maggior parte dei pazienti affetti da ALMS presenta un’intelligenza normale, con normale sviluppo cognitivo, anche se sono stati segnalati alcuni casi con ritardo mentale, difetti del linguaggio, difficoltà nel risolvere problemi e problematiche psichiche. Circa un terzo dei pazienti sembra avere disturbi neurologici.
Genetica
Nel 2002, due differenti gruppi di ricerca hanno identificato il gene ALMS1 e hanno segnalato la presenza di mutazioni specifiche responsabili della sindrome di Alström non osservabili nella popolazione generale. Il gene ALMS1, la cui funzione non è ancora nota, è costituito da 23 esoni e codifica per una proteina di 4.169 aminoacidi che potrebbe svolgere un ruolo importante nella funzione del citoscheletro. Sono note 25 mutazioni di ALMS1. Nell’ALMS è frequentemente osservabile una variabilità fenotipica anche fra individui con la stessa mutazione, probabilmente a causa di altri fattori genetici o ambientali che interferiscono con il gene ALMS1.
Patologia
Le rare valutazioni autoptiche o gli esami istologici di biopsie di tessuti, principalmente fegato e cuore, hanno sistematicamente evidenziato la presenza di una marcata fibrosi interstiziale, sia focale che disseminata. Tuttavia al momento attuale non è chiaro se i processi di fibrosi costituiscano il “primum movens” della sindrome o se siano più semplicemente un epifenomeno.
Diagnosi
La diagnosi di sindrome di Alström non è agevole, per il sovrapporsi di alcuni aspetti clinici di questa patologia con altre malattie. La sindrome è molto rara, poco conosciuta e talvolta i pazienti con ALMS vengono erroneamente considerati affetti da altre patologie più note quali ad esempio la sindrome di Bardet-Biedl, l’amaurosi congenita di Leber, la cardiomiopatia dilatativa sporadica, la sindrome di Usher e alcuni disordini mitocondriali. Oltre al caratteristico fenotipo e correlati clinici, per porre diagnosi di sindrome di Alström è necessario effettuare uno studio del gene ALMS1 per la ricerca di mutazioni.
Terapia
Non sono stati descritti trattamenti specifici per la sindrome di Alström. È necessario un approccio multidisciplinare al paziente, effettuato da specialisti esperti in questa patologia, al fine anche di prevenire le sue molteplici complicanze. La sindrome di Alström non coinvolge solo il paziente, ma anche la sua famiglia alla quale è quindi necessario fornire un adeguato supporto.
Bibliografia essenziale
1.Alstrom CH. et al. Retinal degeneration combined with obesity, diabetes mellitus and neurogenous deafness: a specific syndrome (not hitherto described) distinct from the Laurence-Moon-Bardet-Biedl syndrome: a clinical, endocrinological and genetic examination based on a large pedigree. Acta Psychiatr Neurol Scand Suppl. 1959;129:1-35.
2.Paisey RB, et al. Hypertriglyceridaemia in Alstrom's syndrome: causes and associations in 37 cases. Clin Endocrinol (Oxf). 2004;60:228-31.
3. Collin GB, et al. Mutations in ALMS1 cause obesity, type 2 diabetes and neurosensory degeneration in Alstrom syndrome. Nat Genet. 2002;31:74-8.
4. Quiros-Tejeira RE, et al. Early-onset liver disease complicated with acute liver failure in Alstrom syndrome. Am J Med Genet. 2001;101:9-11
5.Marshall JD, et al. New Alstrom syndrome phenotypes based on the evaluation of 182 cases. Arch Intern Med. 2005;165:675-83.
ultimo aggiornamento: 9 novembre 2007